Joubert syndrome
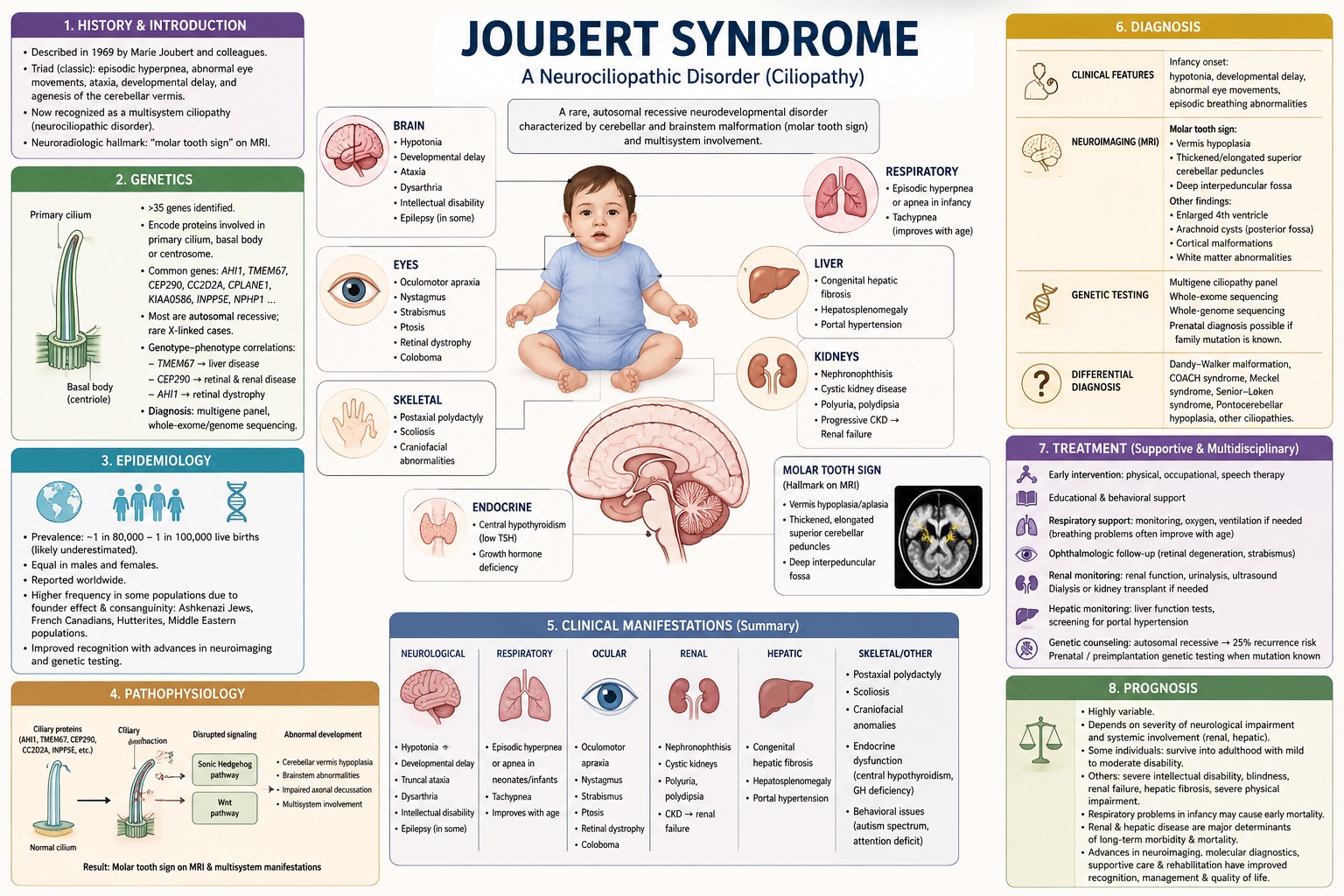
History and Introduction
Joubert syndrome is a rare inherited neurodevelopmental disorder characterized primarily by malformation of the cerebellum and brainstem. The condition was first described in 1969 by the Canadian neurologist Marie Joubert and colleagues, who reported a group of siblings presenting with episodic hyperpnea, abnormal eye movements, ataxia, developmental delay, and agenesis of the cerebellar vermis.
Since the original description, the clinical spectrum of the disorder has expanded considerably, and Joubert syndrome is now recognized as a multisystem disease belonging to the group of disorders known as ciliopathies.
The neuroradiological hallmark finding is the “molar tooth sign” on magnetic resonance imaging (MRI), resulting from hypoplasia or aplasia of the cerebellar vermis, together with characteristic abnormalities of the brainstem and superior cerebellar peduncles. Joubert syndrome, as a neurociliopathic disorder, has genetic and clinical heterogeneity. It means that, in addition to neurological abnormalities, affected individuals may develop multiorgan involvement including the retinal, renal, hepatic, skeletal, and endocrine systems. The clinical presentation varies considerably, from relatively mild developmental deficits to severe multisystem disease.
Genetics
The genes associated with Joubert syndrome encode proteins that are involved in the formation of the primary cilium, the basal body (the foundation of cilia), or the centrosome. These structures are essential for cellular signaling during embryonic development. Commonly pathogenic genes include AHI1, TMEM67, CEP290, CC2D2A, CPLANE1, KIAA0586, INPP5E, and NPHP1.
These gene mutations lead to impaired structure and function of the primary cilia, which disrupts key cellular signaling pathways. These pathways include the Sonic Hedgehog and Wnt pathways, which are critical for normal organogenesis and nervous system development. Joubert syndrome is genetically heterogeneous and is caused by pathogenic variants in more than 35 identified genes.
Most forms of the disease are inherited in an autosomal recessive manner, although rare X-linked cases have also been reported. Certain genotype–phenotype correlations have been reported, for example, TMEM67 mutations are commonly associated with liver disease, CEP290 mutations are frequently linked to retinal and renal involvement, and AHI1 mutations are often associated with retinal dystrophy. Due to the extensive genetic heterogeneity, molecular diagnosis typically relies on multigene panels, whole-exome, or whole-genome sequencing.
Epidemiology
Joubert syndrome is a very rare disorder with an estimated prevalence of approximately 1 in 80,000 to 1 in 100,000 live births. However, the true prevalence is likely underestimated, as mild or atypical cases may go undiagnosed. This condition, with autosomal recessive inheritance, occurs in males and females equally and has been reported worldwide in different populations.
However, increased prevalence has been reported in certain groups due to founder effects and consanguinity, including among Ashkenazi Jewish populations, French Canadians, Hutterites, and some Middle Eastern populations. Advances in neuroimaging and genetic testing have improved the recognition of this disorder, leading to increased identification of milder phenotypes and a broader range of clinical presentations.
Pathophysiology
The pathophysiological mechanism of the syndrome is fundamentally related to cilia dysfunction. Primary cilia are microtubule-based organelles present on nearly all human cells and function as sensory and signaling centers involved in cell differentiation, proliferation, migration, and tissue organization. During the embryonic phase, cilia regulate several critical signaling pathways, including Sonic Hedgehog signaling, which is essential for the patterning of the central nervous system and other organs. Mutations affecting ciliary proteins disrupt cerebellar and brainstem development, neuronal migration, and axonal decussation.
Neuropathological features include hypoplasia or aplasia of the cerebellar vermis, elongation and thickening of the superior cerebellar peduncles, and deepening of the interpeduncular fossa. These changes produce the characteristic molar tooth sign on MRI. Although total or partial absence of decussation of major neural pathways contributes to impaired motor coordination and oculomotor dysfunction. Since primary cilia are also essential for the development and maintenance of other organ systems, ciliary dysfunction additionally affects retinal photoreceptors, renal tubules, and biliary ducts, explaining the multisystem manifestations of the disorder.
Clinical Manifestations
Joubert syndrome exhibits various clinical manifestations, ranging from mild developmental delay to severe involvement of body organs. Neurological manifestations usually begin in infancy and include hypotonia, developmental delay, truncal ataxia, and sometimes epilepsy (the latter is not as common). Epileptic seizures occur in some affected individuals and can manifest in different semiologies. Epileptiform activity arises from a subcortical area and, in some cases, leads to a normal EEG. Many affected individuals exhibit delayed motor milestones, discoordination, dysarthria, and intellectual disability of varying severity.
Breathing abnormalities, characterized by episodic tachypnea or apnea, are most prominent during the neonatal period and infancy and often improve with age. Oculomotor engagement is relatively common and includes oculomotor apraxia, nystagmus, strabismus, and ptosis. Ocular involvement may include retinal dystrophy and coloboma, which can lead to progressive visual impairment or blindness.
Behavioral abnormalities, including autism spectrum disorder and attention deficits, may also occur. which can lead to progressive visual impairment or blindness. Renal disease is an important source of morbidity and may manifest as nephronophthisis, cystic kidney disease, polyuria, polydipsia, and progressive chronic kidney disease leading to renal failure during childhood or adolescence.
On the other hand, hepatic involvement includes congenital hepatic fibrosis, hepatosplenomegaly, and portal hypertension. Postaxial polydactyly, scoliosis, craniofacial abnormalities, and endocrine dysfunction, including central hypothyroidism (with low TSH level) and growth hormone deficiency, are all additional manifestations. The severity of systemic features varies considerably among individuals and is linked to the underlying genetic defect.
Diagnosis
The diagnosis of Joubert syndrome is based on clinical findings and neuroimaging hallmarks; however, it is confirmed by genetic testing. During infancy, affected children present with hypotonia, developmental delay, abnormal eye movements, and episodic dysregulation of breathing. MRI of the brain plays a key role in diagnosis, revealing the molar tooth sign, an indication of cerebellar vermis hypoplasia, as well as thickened, elongated superior cerebellar peduncles and a deep interpeduncular fossa.
Other neuroimaging findings may include enlargement of the fourth ventricle, vermis atrophy/hypoplasia, arachnoidal cysts in the posterior fossa, cortical malformations, and various white matter abnormalities. However, molecular genetic testing is important for confirming diagnosis and identifying genotype–phenotype correlations.
Diagnostic/genetic methods include targeted multigene ciliopathy panels, whole-exome sequencing, and whole-genome sequencing. Prenatal diagnosis may also be possible in families with known pathogenic variants.
Differential diagnosis includes other congenital cerebellar malformations and ciliopathies, including Dandy–Walker malformation, COACH syndrome, Meckel syndrome, Senior–Løken syndrome, and Pontocerebellar hypoplasia.
Treatment
Today, there is no curative treatment for Joubert syndrome, and the management is supportive, symptomatic, and multidisciplinary. However, early interventions, including physical, occupational, and speech therapy, are necessary to optimize developmental milestones and improve function. Educational support and behavioral interventions are required for children with cognitive or behavioral difficulties.
Infants with severe apnea or respiratory insufficiency require supplemental oxygen,respiratory monitoring and assistance, and mechanical ventilation, although breathing difficulties often diminish with age. Ophthalmologic follow-up is essential for monitoring retinal degeneration, visual acuity, and strabismus.
Regular renal surveillance, including renal function tests, urinalysis, and ultrasonography, is also important because renal disease may progress successively before progressing to chronic renal failure. Management of renal failure may require dialysis or kidney transplantation.
Hepatic monitoring should include assessment of liver function and evaluation for portal hypertension in patients at risk for congenital hepatic fibrosis. Genetic counseling is recommended for affected families because most forms are inherited in an autosomal recessive pattern with a recurrence risk of 25% for future pregnancies. Prenatal and preimplantation genetic testing may be available when the causative mutation has been identified.
Prognosis
The variation of prognosis of Joubert syndrome is relatively high and depends largely on the severity of neurological impairment and the coexistence of systemic complications such as renal or hepatic disease. Some individuals with predominantly neurological involvement may survive into adulthood with mild to moderate disability and achieve varying degrees of independence.
Indeed, others experience severe intellectual disability, blindness, progressive renal insufficiency, hepatic fibrosis, and severe physical impairment. Respiratory complications during infancy may contribute to early mortality in severe cases. Renal disease and hepatic complications are among the most important determinants of long-term morbidity and mortality. Advances in neuroimaging techniques, molecular diagnostics, supportive care, and rehabilitation have improved recognition of the disorder and enhanced long-term management and quality of life.